A 50-year-old male with moderate to severe congenital hemophilia A presented to the hemophilia treatment center with chief complaints of bilateral knee and right shoulder pain. The patient denied gastrointestinal (GI) bleeding, fever, night sweats, weight loss, chest pain, and dyspnea. The patient had a baseline factor VIII level of 0-3% and an inhibitor titer of 0 Bethesda units (BU). His right knee was a target joint with additional hemophilic arthropathy issues in his right wrist and right shoulder. Complicated medical history included HIV, hepatitis C, immune thrombocytopenia (s/p splenectomy in 1/1985), mycobacterium avium intracellular infection (MAI), upper GI bleeding, arthritis secondary to hemophilic arthropathy, and chronic back pain due to a fall.
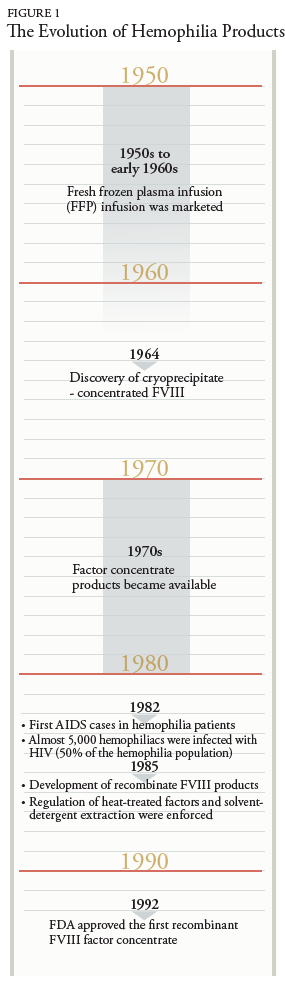
During his infancy, the patient had a prolonged bleeding post-circumcision. The extensive bruising was noted at his 6-month-old checkup, which led to further testing and diagnosis of hemophilia A. His bleeding disorder was initially treated with cryoprecipitate until he was 8 years old. Afterward, the treatment was replaced by on-demand FVIII replacement regimen, which consisted of 3,000 units for mild bleeding and 7,000 units for severe bleeding. In 2013, the patient was switched to prophylaxis therapy due to recurrent hospitalizations from gastrointestinal (GI) bleeding. The admission laboratory tests revealed a significantly low level of factor VIII activity, 22.4%, unremarkable inhibitor assay of 0 titers, and PTT of 32.5 seconds (H). His bleeding history prior to starting prophylaxis FVIII therapy included right iliopsoas hematomas, right elbow and right knee hemarthrosis, GI bleeding, right wrist hemophilic arthropathy/hemarthrosis, right shoulder pain with bicipital tendinopathy, and right hip pain related to iliopsoas bleeding. Upon multiple hospitalizations due to unrevoked GI bleeding, the patient was placed on an escalated prophylaxis dosing of FVIII replacement therapy with Kogenate 3,100 units 3 times weekly.
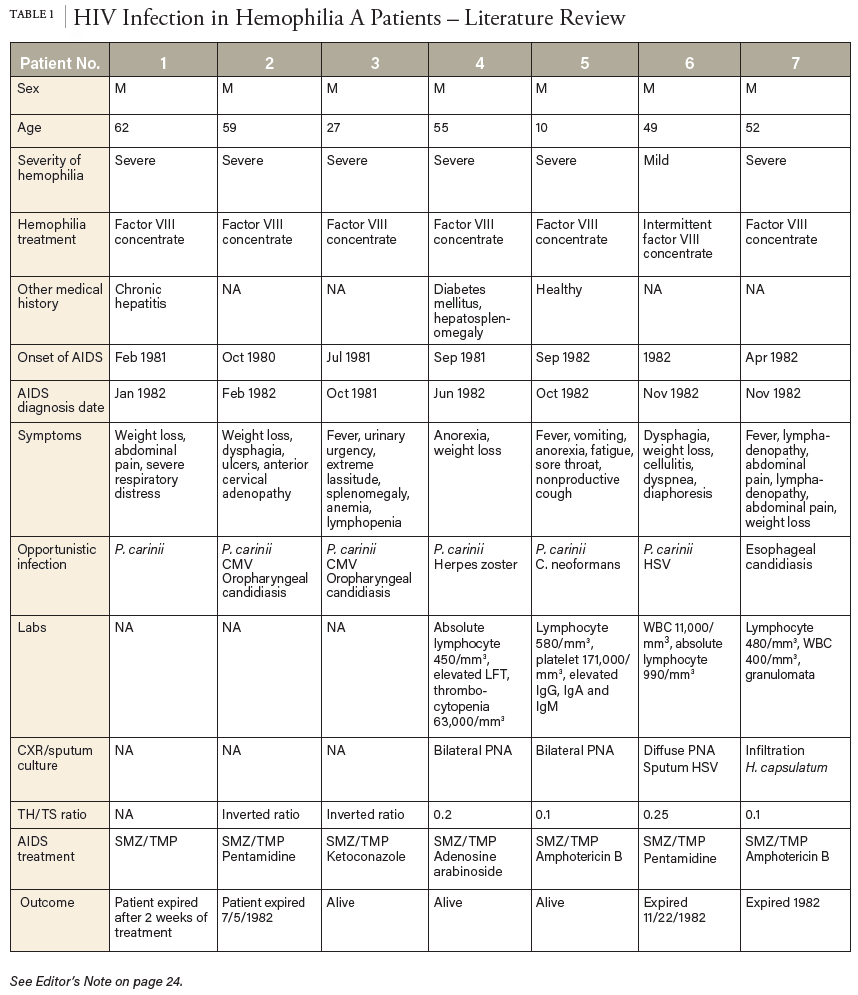
Status post splenectomy due to chronic ITP in 1984, he was diagnosed with acquired HIV and hepatitis C. The patient was treated with Zidovudine (Retrovir) for 9 years, then therapy was discontinued, assumingly due to noncompliance. Ten years later, the patient was rehospitalized due to MAI in the form of a neck mass; he was treated with clarithromycin, ethambutol, and rifabutin. Other readmission complications included candidal esophagitis and AIDS status. Diagnostic laboratory tests showed a critically low CD4/CD8 lymph ratio of 0.65 (L) and a CD4 count of 612/μL. HIV/AIDS treatment was reinitiated with Atripla (efavirenz, emtricitabine, tenofovir) and later changed to Odefsey (emtricitabine, rilpivirine, tenofovir) (serum creatinine 0.84mg/dL, eGFR >60 mL/min/1.73m2, ALT/AST 15 and 23U/L, respectively). His HIV viral load of 3970/mL had been intermittently detectable since 2012, while his hepatitis C viral load had been undetectable since his hospitalization in 2013.